Genetika
Genetická péče zahrnuje následující postupy
1. Určení přesné genetické diagnózy
Jejím cílem je určení klinické jednotky. Přesný typ muskulární dystrofie je odhalen buď na základě DNA analýzy záchytem příčinné mutace, či na základě muskulární biopsie s prokázaným deficitem daného proteinu-produktu tohoto genu (např. dystrofinového proteinu).
2. Určení typu dědičnosti
Jejím cílem je stanovení, kdo má v rodině riziko, že onemocní on sám, nebo jeho potomci a jaké toto riziko je.
3. Nabídnutí možností genetické prevence
Jedná se nabídnutí možnosti prenatální genetické diagnostiky, eventuelně preimplantační genetické diagnostiky a podání informací o možných technikách a jejich rizicích.
1. Diferenciální diagnostika muskulárních dystrofií
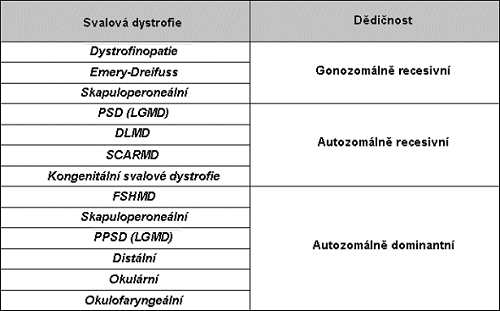
DMD/BMD - Duchenenova-Beckerova progresivní svalová dystrofiePSD - pletencová svalová dystrofie,LGMD - " limb girdle muscular dystrophy"DLMD - "Duchenne like muscular dystrophy" , dětský typ připomínající Duchenneovu formuSCARMD - "severe childhood muscular dystrophy - těžká autosomálně recesivní muskulární dystrofie dětského věkuFSHMD - facioskapulohumerální muskulární dystrofie
Nejčastější muskulární dystrofie jsou dystrofinopatie, které se kliniky projevují čtyřmi samostatnými klinickými jednotkami:
- Duchenova muskulární dystrofie (incidence 1: 3500 u mužů)
- Beckerova muskulrání dystrofie (incidence 1:25000 u mužů)
- syndrom crampi, myalgia a myoglubinuria (bolestivé křeče, bolesti a přítomnost myoglobinu v moči) (vzácné)
- dilatační kardiomyopatie (vzácné)
2. Typy dědičnosti u muskulární dystrofiíGenetická rizika u gonozomálně recesivní dědičnosti
Dědičnost dystrofinopatií je gonozomálně recesivní. Choroba se vyskytuje prakticky u mužů a v rodinách se přenáší přes fenotypicky zdravé ženy přenašečky. U přenašeček se mohou vyskytovat některé klinické či laboratorní projevy choroby, tyto ženy se označují jako manifestní přenašečky. Ze všech přenašeček je manifestních přenašeček 19%. Čerstvé mutace tvoří u DMD 1/3 případů, u BMD jsou vzácnější. V případě čerstvé mutace u postiženého chlapce nemá matka zvýšené riziko pro další potomky.Genetická rizika pro matku postiženého s DMD:
- Žena, která má postiženého syna a dalšího postiženého příbuzného po mateřské linii, je jistá (obligatorní) přenašečka. Žena přenašečka má pro své syny 50% riziko postižení a pro dcery pravděpodobnost přenašečství 50%. Sestra jisté přenašečky má pravděpodobnost přenašečství 1/2, pro své syny má riziko postižení 1/4 a pro dcery pravděpodobnost přenašečství 1/4.
-
Je-li v rodině postižen jen jeden, může se u něho jednat o čerstvou mutaci (pravděpodobnost 1/3) nebo je jeho matka přenašečka. Matka jediného postiženého v rodině má tedy pravděpodobnost přenašečství 2/3, pro své syny má riziko postižení 1/3 a pro dcery 1/3 přenašečství. Je-li u matky jediného postiženého detekováno přenašečtví (například záchytem mutace), pak se u ní mohlo jednat o čerstvou mutaci, nebo zdědila mutaci od své matky či otce (u kterého vznikla čerstvá mutace v zárodečné buňce). V případě čerstvé mutace u matky nemá její sestra ani další ženské příbuzné zvýšené riziko pro své potomstvo. Není-li u matky sporadického případu detekována kauzální mutace, která u jejího postiženého syna byla detekována, pak:
-
buď není přenašečka (čerstvá mutace vznikla u jejího syna) a riziko pro další děti je pak nezvýšeno,
-
nebo mutace je přítomna pouze v některých buňkách mateřského těla (somatický mozaicismus) a může, ale nemusí být detekovatelná v bílých krvinkách, ze kterých se běžně DNA izoluje. Nebo je mutace u matky přítomna pouze v zárodečných buňkách (germinální mozaicismus) a není pak detekovatelná v bílých krvinkách.
-
U DMD se uvádí, že až germinální mozaicismus má 14% (v různé literatuře 10-20%) přenašeček, ženy s germinálním mozaicismem mají samozřejmě zvýšené riziko pro potomstvo, výši tohoto rizika však upřesnit nelze.
Genetická rizika pro sestru postiženého s DMD: Riziko pro potomstvo sestry postiženého závisí na tom, zda matka je či není přenašečka. Je-li matka postiženého chlapce přenašečka, jeho sestra má pravděpodobnost přenašečství 1/2 a tedy pro své syny pravděpodobnost postižení 1/4. S každým poklesem příbuznosti je riziko poloviční.Genetická rizika pro potomky postiženého:Postižený gonozomálně recesivní chorobou má všechny syny zdravé a všechny dcery přenašečky.Druhé nejčastější muskulární dystrofie - pletencové (LGMD) jsou v 90% autozomálně recesivní a 10% autozomálně dominantní.
Genetická rizika u autozomálně recesivních chorob
Jedinec postižený autozomálně recesivní chorobou je recesivní homozygot, nositel dvou mutovaných alel. Postižení jedinci se zpravidla vyskytují pouze v jedné rodině - všichni jsou potomci stejných rodičů.Genetická rizika pro rodiče postiženého autozomálně recesivní muskulární dystrofiíRodiče postiženého jsou zdraví heterozygoti - přenašeči mutace. Heterozygoti jsou nosiči jedné mutace, druhá alela je normální, označovaná jako standardní. Pro své společné potomky mají 25% riziko postižení. Pokud bude mít rodič potomstvo s jiným partnerem a tento s ním není pokrevně příbuzný, pak genetická prognóza pro potomstvo přenašečů je příznivá.Genetická rizika pro potomstvo postižených jedinců autozomálně recesivní svalovou dystrofiíVšichni potomci postiženého jedince jsou s největší pravděpodobností zdraví nosiči mutace-heterozygoté. Genetická prognóza pro potomstvo postiženého je příznivá.
Genetická rizika u autozomálně dominantních chorob
U autozomálně dominantní choroby je postižený zpravidla nosič jedné mutace - heterozygot. Choroba se většinou přenáší po více generací.Genetická rizika pro rodiče postiženého autozomálně dominantní svalovou dystrofiíVětšina pacientů s autozomálně dominantní chorobou má postiženého rodiče. Postižený rodič má pro své potomstvo 50% riziko. Příznaky u postižených členů téže rodiny mohou být variabilní (variabilní expresivita). Dokonce se může stát, že nosič mutace nemá žádné projevy onemocnění, jev je označován jako neúplná penetrance. Je proto nutno počítat s tím, že pokud jsou oba rodiče zdraví, neznamená to, že pro své děti nemají zvýšené riziko.Genetická rizika pro potomstvo probandaPostižený jedinec své potomstvo 50% riziko.
3. Vyšetřovací postupy u muskulárních dystrofií
3.1. Klinické vyšetření
V prvé řadě je nezbytné podrobné klinické vyšetření pacienta pediatrem/obvodním lékařem, následně neurologem. Opírá se o zhodnocení klinického stavu, posuzuje se objektivní nález a subjektivní potíže pacienta. Důležitými kriterii jsou predominantní postižení svalových skupin, stupeň svalové síly, schopnost chůze, chůze po špičkách a patách, chůze do schodů, zda pacient svede dřep a vztyk, může dát paže nad hlavu, schopnost mimického svalstva atd.V rámci klinického vyšetření je nesmírně důležitá podrobná rodinná anamnéza. Má-li osoba suspektní ze svalového onemocnění postiženého ještě dalšího příbuzného, je důležité posoudit tzv. informativitu rodiny. Za informativní považujeme takovou rodinu, kde výskyt postižení koreluje s určitým typem dědičnosti. Například u gonozomálně recesivní dědičnosti jsou postižení muži v příbuzenském poměru přes ženy přenašečky, nikdy nejsou příbuzní po mužské linii. Postižený muž nemůže mít postiženého syna. Ženy přenašečky jsou většinou zdravé, pokud mají nějaké projevy onemocnění, pak ve srovnání s postiženými muži jsou zpravidla minimální.U autozomálně recesivní dědičnosti jsou postižení jedinci zpravidla vlastní sourozenci, jejichž rodiče jsou zdraví přenašeči. U autozomálně dominantní dědičnosti se choroba zpravidla přenáší z generace na generaci. Zdraví příslušníci rodiny mají zpravidla zdravé děti. Informativitu rodiny nejlépe posoudí klinický genetik.Odpovídá-li rodina určitému typu dědičnost, či vylučuje-li určitý typ genetického přenosu, je to důležité kriterium pro zaměření dalších laboratorních, zejména molekulárně genetických testů.
3.2. Laboratorní vyšetření
3.2.1. Biochemické, neurofyziologické
Pro svalovou dystrofii svědčí zejména zvýšená hladina sérové kreatinkinázy, která může u DMD dosahovat 100 i vícenásobných hodnot . Z laboratorních vyšetření je velmi důležitě elektromyografické vyšetření, suspektní je zejména nález primární svalové léze. Biochemické i neurofyziologické vyšetření jsou pro diagnostiku přínosná, ale k bližší specifikaci typu svalové dystrofie nepostačující. Ke specifikaci typu svalové dystrofie zpravidla přispívají metody molekulárně biologické - analýza DNA ev. mRNA.
3.2.2. Vyšetření metodami molekulárně biologickými
3.2.2.1 DNA analýza delecí jednotlivých exonů DMD/BMD genu
Je-li podezření na muskulární dystrofii, která odpovídá proximálnímu typu a jedná-li se o postiženého mužského pohlaví, či rodina je informativní pro gonozomálně recesivní typ genetického přenosu, je indikována DNA analýza DMD/BMD genu.
3.2.2.1.1. Mutace je u pacienta zachycena.
Tím je diagnóza dystrofinopatie potvrzena a není nutný žádný další diagnostický postup. Pokud vyšetření neindikoval klinický genetik, pak by rodina měla být doporučena na genetickou konzultaci, aby byl zajištěn erudovaný genetický servis. Následně je indikována DNA analýza DMD/BMD genu u matky probanda.
Mutace je u matky postiženého prokázána
Pokud je mutace u matky zachycena, pak je potvrzeno přenašečství dystrofinopatie a tato žena má pro své syny 50% riziko postižení a pro dcery 50% riziko přenašečství. Mutaci buď tato žena zdědila od své matky, nebo u ní došlo k čerstvé mutaci, mutaci také mohla zdědit od svého otce, který má germinální mozaisismus. Následně je indikována DNA analýza u babičky (matky matky). Pokud je u ní mutace prokázána, pak všechny její dcery jsou v riziku přenašečství a samozřejmě další její ženské příbuzné v první lini (matka a sestry). Pokud u babičky není mutace prokázána, pro jistotu je indikována DNA analýza u jejích dcer (sester matky pacienta), neboť babička může mít germinální mozaicismus, který se vyskytuje u 14% přenašeček DMD.
Mutace není u matky postiženého zachycena
Pokud u matky pacienta mutace není prokázána, pak je největší pravděpodobnost, že u syna došlo k čerstvé mutaci a matka přenašečka není. Matka však může mít mutaci pouze v germinálních buňkách (udáváno u 14 % přenašeček) a tedy v DNA izolované ze somatických buněk (bílých krvinek) ji detekovat nemůžeme. Matka, u které nebyla kauzální mutace přítomná u syna detekována, má tedy 7% riziko, že syn bude postižen a dcera přenašečka. Proto doporučujeme i u žen, u kterých nebyla mutace z DNA izolované z periferní krve prokázána, v následných graviditách prenatální diagnostiku.
3.2.2.1.2. Mutace není u pacienta zachycena
Je-li vyšetření na přítomnost delece jednotlivých exonů DMD/BMD genu u pacienta negativní, jsou dvě možnosti. Buď je pacient nosič jiné mutace v DMD/BMD genu nebo se jedná o jinou diagnózu způsobenou mutací jiného než dystrofinového genu. Vzhledem k tomu, že, jak již bylo řečeno, jsou svalové dystrofie heterogenní skupinou onemocnění způsobené mutacemi různých proteinů, k další specifikaci je nezbytná svalová biopsie.
3.2.2.2 Vyšetření muskulární biopsie
Provádí se kompletní vyšetření histologické, histochemické, imunohistochemické a imunofluorescenční. Pro specifikaci typu muskulární dystrofie je klíčové imunohistochemické a imunofluorescenční vyšetření, tato vyšetření nejsou součástí rutinního spektra vyšetření, ale přistupuje se k němu až na základě patologického nálezu v základních barveních a histochemických reakcích.
Pro diagnostiku dystrofinopatií a jim příbuzných svalových dystrofií se diagnóza opírá o detekci proteinů dystrofinového glykoproteinového komplexu (DGC). Používají se protilátky proti proteinům DGC. U dystrofinu se používají protilátky proti tyčinkové doméně dystrofinu (DYS1, aminokyseliny 1181-1388), C- terminální doméně (DYS2, aminokyseliny 11255-11296), a N-terminální doméně (DYS3, aminokyseliny 308-351). Deficience výše uvedených domén odpovídá klinickému obrazu DMD či BMD.
Dále se využívají protilátky proti následujícím proteinům: kalpain 3- odpovídá LGMD2A, dysferlin (LGMD2B), alfa-sarkoglykan (adhalin-LGMD2D), beta-sarkoglykan (LGMD2)E, gama-sarkoglykan (LGMD2C), delta-sarkoglykan (LGMD2F), kaveolin 3 (LGMD1C). Další proteiny nepatřící mezi proteiny dystrofinového glykoproteinového komplexu : emerin, způsobuje X-vázanou formu Emeryho-Dreifussovy svalové dystrofie, merosin (alfa2-laminin) zodovědný za merosinovou kongenitální muskulární dystrofií, spektrin (chybí na nekrotických vláknech, je přítomen na vláknech s deficitním dystrofinem), utrofin-N, utrofin-C- za normálních okolností se exprimuje pouze v oblasti neuromuskulráního spojení, u dystrofinopatií dochází k jeho upregulaci (zvýšené expresi) na povrchu svalových vláken.
mRNA analýza muskulární biopsie
Pokud výsledek muskulární biopsie podporuje diagnózu dystrofinopatie, chybí, či jsou oslabeny jednotlivé domény dystrofinu a je upregulován utrofin, je následně indikována mRNA analýza dystrofinového genu. Rutinně se provádí vyšetření na detekci možných delecí, duplikací a nonsense mutací (mutací způsobujících předčasné ukončení translace). Je-li v mRNA ze svalové biopsie mutace prokázána, je indikováno její potvrzení z genomové DNA. Pokud je mutace rovněž zachycena v genomové DNA, následuje DNA analýza matky a dalších příbuzných za účelem detekce přenašeček v rodině.
Pokud není v mRNA ze svalové biopsie mutace DMD/BMD genu zachycena, je možno, že pacient mutaci v DMD/BMD genu má, tato však nezpůsobuje předčasné ukončení translace. V tomto případě indikujeme sekvenční analýzu DMD/BMD genu z genomové DNA. Je potřeba zdůraznit, že se sekvenční analýza DMD/BMD genu je z mnoha hledisek velmi náročné vyšetření, jehož kapacity v ČR nejsou zcela optimální. Jestliže mutace v DMD/BMD genu je zachycena, indikujeme DNA analýzu na tuto mutaci u matky, následně dalších žen-suspektních přenašeček.
Pokud mutace v DMD/BMD genu není u pacienta prokázána a klinicky obraz a výsledek muskulární biopsie pro dystrofinopatii svědčí, pak se může jednat o dvě možnosti.
1. U pacienta je defekt dystrofinu ve svalu sekundární, kauzální mutace se týká jiného proteinu, který je s dystrofinem v úzkém funkčním propojení a deficience tohoto primárního proteinu, která nebyla svalovou biopsií potvrzena, způsobuje deficit dystrofinu sekundární.
2. Pacient má vzácný typ mutace, který nebyl sekvenční DNA analýzou dystrofinového genu odhalen, tento případ je však výjimečný. V obou případech nemáme přesnou diagnózu, nemůžeme tedy ani stanovit přesný typ dědičnosti a nemůžeme rodině nabídnout žádná opatření.Prokáže-li se v muskulární biopsii deficince jiného proteinu než dystrofinu, jsou další postupy obdobné. Jedná se o mRNA a následně DNA analýzu příslušného proteinu. Pro některé proteiny se však mRNA, ani DNA analýza u nás rutinně neprovádí.
Pro lepší orientaci uvádíme rekapitulaci diagnostického procesu v přehledu:
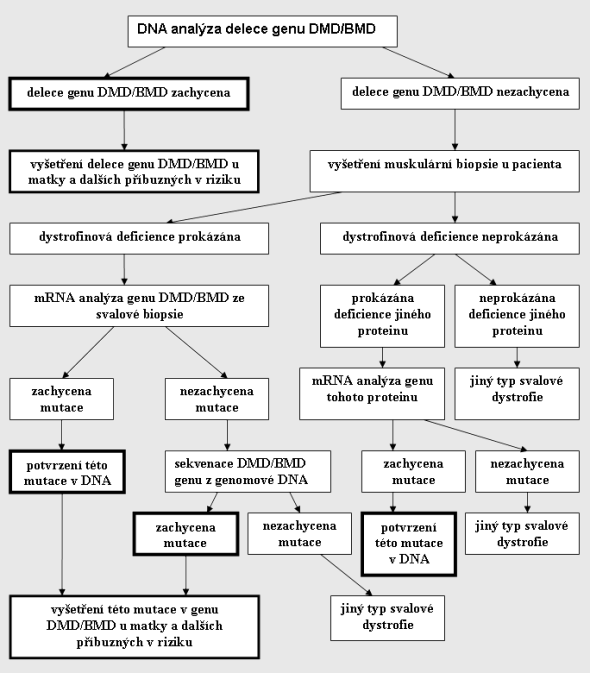
Není-li podezření na dystrofinopatii, je indikována přímo muskulární biopsie a následující postupy jsou obdobné. Tučné jsou znázorněny závěry vedoucí k určení přesného typu muskulárná dystrofie.
3.3. Vyšetření rizikových členů rodiny
U dystrofinopatie je v souladu s gonosomálně recesivní dědičnosti postupováno v ženské linii - vyšetření matky, sester, sester matky babičky, atd.Pokud je u probanda prokázána mutace, pak je situace jednoduchá.
U matky je indikována DNA analýza na tuto mutaci. Pokud je výsledek pozitivní - mutace je zachycena, pak se vyšetří sestry probanda , babička a dále s pokračuje v ženské linii.
Pokud je u matky vyšetření negativní, pak její sestry nemohou mít mutaci, ale sestry probanda mohou, jelikož matka může mít germinální mozaicismus (udáváno cca ve 14%)Jestliže u probanda mutace není prokázána a je vysoká pravděpodobnost, že se u něho jedná primárně o dystrofinopatii, je indikována nepřímá, haplotypová analýza rodiny využívající intragenové DNA polymorfismy. Polymorfní alely, které má postižený proband, jsou označeny za rizikový haplotyp.
Pokud je více postižených v rodině, pak by samozřejmě všichni měli mít tento rizikový haplotyp. Nevýhoda této nepřímé (haplotypové) analýzy spočívá v určitých limitacích. První je, že pacient může mít mutaci jiného kauzálního genu. Druhá limitace nastává v případě, kdy sice kauzální mutace v dystrofinovém genu je přítomna, ale v důsledku rekombinace dojde k výměně DNA materiálu mezi zdravým a postiženým chromozomem a pak interpretace výsledku je složitá a záleží na konkrétním výsledku.
3.4. Preventivní opatření - prenatální diagnostika
Prenatální diagnostika se opírá o metody invazivní a neinvazivní. Invazivní metody spočívají v odběru materiálu plodu a jeho následnému vyšetření chromozomálnímu, DNA či biochemickému. Metody invazivní prenatální diagtnostiky je odběr choriové biopsie, plodové vody, placenty, krve z pupečníku. Tyto se standardně v ČR provádějí. Odběr muskulární biopsie u plodu se u nás rutinně neprovádí. Neinvazivní prenatální diagnostika jsou všechna ostatní neinvazivní vyšetření. Jedná se například u ultrazvukové vyšetření, biochemické screeningy matky a další: Pro problematiku svalových dystrofií neinvazivní metody prenatální diagnostiky nejsou přínosné vůbec, nebo jsou jen orientační (napři určení pohlaví plodu z ultrazvukového vyšetření).
Invazivní prenatální diagnostika může být indikována u rizikových gravidit přesahujících 1%. Z indikace dystrofinopatie se provádí u těch gravidit, kde má žena zvýšené riziko, že je přenašečka mutace. Podmínkou je, že v rodině je buď detekována kauzální mutace, nebo má postižený proband výsledek biopsie svědčící pro dystrofinopatii a následné vyšetření nepřímou DNA analýzou v rodině je informativní. Samozřejmě preferujeme situace, kdy je potvrzena kauzální mutace a děláme všechno proto, abychom ji potvrdili. Ne vždy však máme v rodině příčinnou mutaci detekovánu a zejména při běžící graviditě není pak možno z časových důvodů sekvenční analýzu DMD/BMD genu dokončit. Proto je důležité, aby rodiny s genetickým pracovištěm dobře spolupracovali a pokud u nich genetické vyšetření dosud neproběhlo, si jej vyžádali.
Snahou je, aby prenatální diagnostika byla co nejdříve. Proto většina pracovišť indikuje vyšetření choriové biopsie. Z materiálu se izoluje DNA plodu. Nejprve se rychle vyšetří pohlaví plodu (nejčastěji metodami DNA analýzy na některý z genů na Y chromozomu). Pokud se potvrdí, že plod je mužského pohlaví, v zápětí proběhne DNA analýza DMD/BMD genu - na kauzální mutace, či přítomnost rizikového haplotypu. Těhotná žena má samozřejmě možnost nabízenou prenatální diagnostiku odmítnout, či v jakémkoliv stádiu vyšetření od vyšetření odstoupit bez jakýchkoliv důsledků. Pokud je potvrzeno, že plod mužského pohlaví je nosič mutace, je nabídnuto přerušení gravidity z genetické indikace. Z genetické indikace se může přerušit gravidita do 24. týdne těhotenství. V žádném případě nesmí být žena do přerušení těhotenství nucena.
Protože je pro mnoho žen přerušení gravidity nepřijatelné vůbec nebo psychicky enormně zatěžující, je jimi často vyžadována tzv. preimplantační diagnostika. Preimplantační diagnostika znamená oplození mimo tělo matky (in vitro fertilizace-IVF), následná genetická analýza buněk získaných embryí a transfer pouze zdravých embryí do dělohy ženy. V ČR se rutinně provádí preimplantační diagnostika na pohlaví plodu, kde v rodinách s rizikem gonozomálně recesivní choroby se následně do dělohy transferují pouze embrya ženského pohlaví. Jsou pracoviště asistované reprodukce, kde již i u nás byla provedena preimplantační diagnostika na přítomnost mutace DMD/BMD genu u plodu mužského pohlaví.
4. Závěr
Výše uvedené poznámky jsou určeny k základní orientaci pro rodiny s výskytem dystrofinopatie a rozhodně nenahrazují genetickou konzultaci. Pokud jsou některé údaje nesrozumitelné, omlouvám se.
Další informace včetně grafických schemat můžete dostat na: http://www.eurogentest.org/patient/info/public/unit6/patients_czech.xhtml